Predictive toxicology is a rapidly evolving field that uses in silico—or computational—tools to forecast the potential toxicity of chemicals on living organisms and ecosystems. The field is driven by the need to assess the safety of an increasing number of existing and new compounds—including drugs, industrial chemicals, cosmetics, and environmental agents—in a way that is faster, cheaper, more informative, and more ethical than animal testing.
Read-across
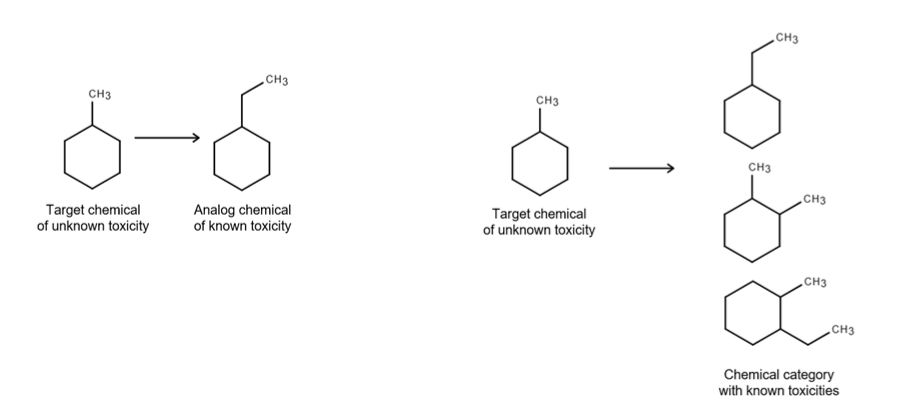
Read-across is a fundamental and widely used technique to predict the hazardous effects of exposure to data-poor chemicals. It is conducted by drawing comparisons to other chemicals (known as analogues) or groups of chemicals (referred to as categories) to make a hazard determination for the chemical under review (the target chemical). The advantage of read-across is that a chemical without data on a specific hazard endpoint can be compared to another chemical or category of chemicals for which data already exist, obviating the need for additional testing.
There are two main approaches to read-across (OECD, 2025). In the analogue approach, the target chemical, for which there will be little or no hazard endpoint data, is compared to a single other chemical or a small grouping of chemicals. The category approach is based on grouping closely related chemicals into a category, rather than treating them as individual chemicals.
Often, chemicals are grouped when they undergo (bio)transformation to common compounds and/or are different compounds that have qualitatively similar properties (ECHA, 2017). A read-across proposal must be justified adequately for each endpoint, and what constitutes sufficient justification for read-across may include structural and mechanistic similarities (ECHA (2017), OECD (2025), EFSA (2025)).
QSAR
While read-across relies on experimental data from structurally and/or mechanistically similar chemicals, Quantitative Structure–Activity Relationship (QSAR) models use a mathematical model to predict a chemical’s toxicity based on information about its chemical structure. In other words, a QSAR model evaluates molecular descriptors, such as a chemical’s molecular weight, polar surface area, lipophilicity, or number of aromatic rings, and applies a mathematical or statistical model to predict a biological activity, such as toxicity. There are various types of QSAR models, including mechanistic (e.g., based on the adverse outcome pathway for skin sensitisation), statistical (e.g., multiple linear regression or random forests), hybrid (combine mechanistic and statistical), rule-based expert systems, and machine-learning/AI-driven.

Select model developers
- US Environmental Protection Agency Computational Toxicology and Exposure Online Resources | Website
- US Environmental Protection Agency Predictive Methods to Assess Hazard under TSCA | Website
- US NTP Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) | Website
- OECD QSAR ToolBox | Website
- Danish QSAR Database | Website
- ACD Percepta | Website
- ToxTree | Website
- Lhasa Limited | Website
- MultiCASE, Inc. | Website
- Simulations Plus | Website
- KREATiS | Website
Resources
- PETA Science Consortium International Webinar Series on the Use of In Silico Tools in Toxicology | Website
- PETA Science Consortium International factsheets on in silico tools for predicting various endpoints | Website
- US Environmental Protection Agency Computational Toxicology and Exposure Online Resources | Website
- OECD. (Q)SAR Assessment Framework: Guidance for the regulatory assessment of (Quantitative) Structure Activity Relationship models and predictions. 2024. Second Edition. https://doi.org/10.1787/bbdac345-en
- OECD. Integrated Approaches to Testing and Assessment (IATA) Case Studies Project. https://www.oecd.org/en/topics/sub-issues/assessment-of-chemicals/integrated-approaches-to-testing-and-assessment.html
- OECD. Guidance on Grouping of Chemicals. 2025. Third Edition, OECD Series on Testing and Assessment, No. 418, OECD Publishing, Paris. https://one.oecd.org/document/ENV/CBC/MONO(2025)19/en/pdf
- ECHA. Read-Across Assessment Framework (RAAF). 2017. https://echa.europa.eu/documents/10162/13628/raaf_en.pdf/614e5d61-891d-4154-8a47-87efebd1851a
- EFSA. Guidance on the use of read‐across for chemical safety assessment in food and feed. 2025. https://doi.org/10.2903/j.efsa.2025.9586
- Scientific Committee on Consumer Safety (SCCS). Memorandum on the use of in silico methods for assessment of chemical hazard. 2016. https://health.ec.europa.eu/publications/memorandum-use-silico-methods-assessment-chemical-hazards_en
- Collaborative Adverse Outcome Pathway Wiki (AOP-Wiki). https://aopwiki.org/
- Wall J, Sayre R, Smith D, et al. Development of the toxicity values database, ToxValDB: a curated resource for experimental and derived human health-relevant toxicity data. Computational Toxicology. 2025;35:100365. https://doi.org/10.1016/j.comtox.2025.100365
- Lou S, Yu Z, Huang Z, et al. In silico prediction of chemical acute dermal toxicity using explainable machine learning methods. Chemical Research in Toxicology. 2024;37(3):513-524. https://doi.org/10.1021/acs.chemrestox.4c00012
- Carusi A, Sanchez Dorado J, Sözüdoğru E. Adverse Outcome Pathway – Study Report. Wittwehr, C. and Whelan, M. editor(s), EUR 30925 EN, Publications Office of the European Union, Luxembourg, 2022, ISBN 978-92-76-45228-7. https://data.europa.eu/doi/10.2760/476003.
- Moustakas H, Date MS, Kumar M, et al. An end point-specific framework for read-across analog selection for human health effects. Chem Res Toxicol. 2022;35(12):2324-2334. https://doi.org/10.1021/acs.chemrestox.2c00286
- Lancia P, Louazzani M, Gros L, et al. Overview of in silico tools to evaluate human health toxicity, ecotoxicity, and toxicokinetic profiles in the hazard assessment of chemicals used in cosmetics. Chem Res Toxicol. 2025;38(10):1652-1680. https://doi.org/10.1021/acs.chemrestox.4c00534